
Remember me









As of 2024, multiple sclerosis remains a clinical diagnosis, and the disease has been traditionally characterized principally according to its clinical phenomenology. Such phenotyping is inherently defined by what can be observed and measured. The field of MS is on the precipice of change, poised to re-conceptualize the disease based on recent advances in bench science, biomarkers, imaging techniques, and ever-more sensitive clinical tools with profound implications for how we diagnose MS, determine pathways for intervention, measure outcomes, and ultimately care for people living with the disease.
It is now relatively well accepted that MS is characterized by both acute and diffuse inflammation as well as neurodegeneration, all of which begin from the onset and occur to varying degrees throughout the disease course. Still, we have anchored to relatively broad clinical frameworks [1], even as we have gained a more nuanced understanding of the immunopathologic underpinnings of disease. Given this, there has been a recent call in the MS literature to move toward a biologic/mechanistic view of MS as a spectrum of disease [2▪▪,3▪▪]. While clinical phenotyping was essential to organizing the pivotal clinical trials that rendered MS a treatable disease 30 years ago [4▪,5], the field is now grappling with how classifying patient cohorts by clinical phenotype alone may obscure the diverse biologic, pathologic, and modulatory factors that underlie the spectrum of disease and its heterogeneous outcomes.
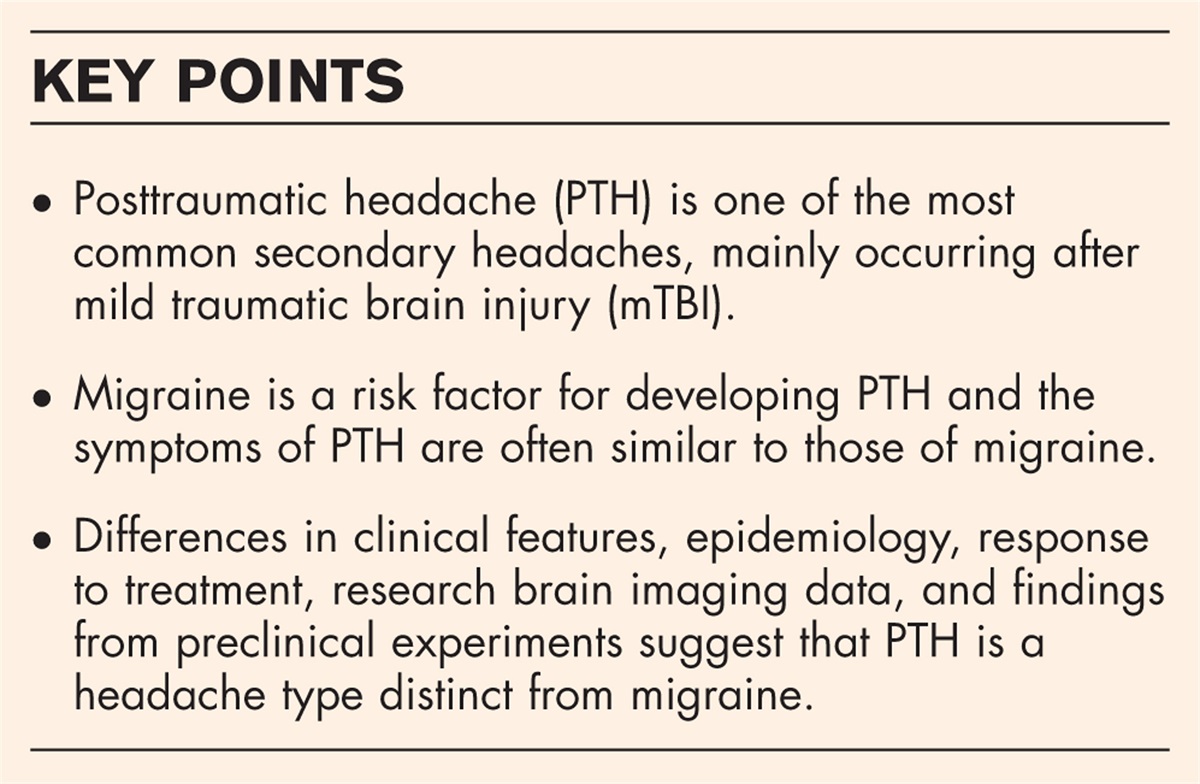
 Box 1:
Box 1: no caption available
LEVELS OF EXPLANATION AND TERMINOLOGYWith the emerging approach to MS as a spectrum of disease predicated not purely on clinical observation but on mechanistic underpinnings, the field has entered a period of conceptual disarray characterized by a tangle of terminology that form a barrier to consensus. The lack of definitional alignment for conceptualizing MS as a spectrum of disease can be addressed by clarifying the different levels of explanation of the disease – namely, heterogenous manifestations of the clinical course versus the diverse biological underpinnings of disease progression.
In this Current Opinion, we seek to define and explicate these various levels of explanation through which MS as a disease spectrum are being evaluated and understood. A related goal of this review is to clarify at which level of explanation the current terminology of MS research operates, to provide a cogent guide to the lexicon of MS inquiry.
Several recent publications center the mechanistic underpinnings of MS as the “ground truth” of the disease [3▪▪,6]. In this review, we will begin with a discussion of this emerging ground truth, and work our way up through sequential levels of explanation at increasing levels of remove from the cellular mechanisms into realms of inference and abstraction.
We will use as our framing mechanism the concept of disease manifestations occurring above and below the clinical threshold of detection. The notion that MS is characterized by a dynamic clinical threshold is an essential component of the topographical model of MS, a unified depiction of disease course which visualized MS as a spectrum characterized by the admixture of inflammation and neurodegeneration across the disease continuum [7] (see Fig. 2). Importantly, this model incorporates the role of lesion localization (disease topography) and the extent of compensatory functional reserve, two increasingly recognized drivers of disability accrual [8,9] that were previously absent from traditional clinical course depictions.
As a unified model of MS disease course, the topographical model helped to encapsulate drivers of disease heterogeneity, resolve the clinical-radiological paradox by distinguishing above- and below-threshold disease burden [9], and visualize the dynamic transition to clinically-apparent progression. This model was designed as a clinical manifestation framework based on traditional MS clinical assessments and conventional MRI findings, and did not incorporate cellular mechanisms or the role of emerging biomarkers. In this review, we will use the topographical model framework of above- and below-threshold manifestations of the disease, and integrate the current understanding of mechanistic drivers of MS across the disease continuum (Table 1).
 Table 1:
Table 1: MS as a disease spectrum: seven levels of explanation
 Table 1(Continued):
Table 1(Continued): MS as a disease spectrum: seven levels of explanation
1. “Ground truth”: cellular mechanisms and immunopathogenesisSeveral recent publications have called for consideration of an immunopathologic and mechanism-driven framework for characterizing MS [2▪▪,3▪▪,10]. The International Committee on Clinical Trials in Multiple Sclerosis (Kuhlmann et al.) suggest that it is time to move from the traditional clinically-based definitions to a biologically-based understanding of disease progression. At the root of their recommendation are data that demonstrate that several concurrent mechanisms drive immunopathologic tissue injury and progression in MS [2▪▪].
The evolving understanding of cellular mechanisms in MS has moved beyond that of inciting inflammatory influx from the periphery to focus on the presence of CNS-resident (compartmentalized) inflammation observed throughout the disease course. In a review of these pathophysiological targets, Yong and Yong frame these contributions of acute and chronically active inflammation along with the mismatch of reparative and remyelinating mechanisms [11▪]. They describe that the differences between pathologic changes early in the disease continuum versus late is now understood to be incremental and qualitative in nature. This view of disease evolution acknowledges a shift from acute macroinflammatory injury seen with focal lesions to widespread chronic inflammation and neurodegeneration marked by a failure of remyelination and repair. This perspective disrupts a dichotomized view of distinct disease phases in favor of a combination of interdependent and dynamic pathobiological axes [12–15].
Seeking greater precision in MS classification, Pitt et al. further the notion of multiple pathobiological axes to put forth an example of how vastly different pathologic profiles can drive individual patients to present with the same clinically-apparent features [3▪▪].
It has long been recognized that pathological heterogeneity could help to explain diverse MS clinical manifestations, disease severity, and treatment response; Pitt et al. emphasize how apparent clinical homogeneity could obscure such crucial differences in underlying mechanistic processes (see Fig. 1). This concept underscores how clinical phenotype may not track with “ground truth” drivers of disease, and greater precision in categorization based on pathologic variability is warranted. As the authors point out, a unique mix of genetic, biological, and environmental factors are present in any given individual at a given time point, yielding a great many potential combinations of modifiers that contribute to a spectrum of disease across pathologic (and reparative) axes. Clinical trial design incorporating such biological descriptors will improve outcome generation, especially in progressive MS, where a key current limitation is the lack of primary outcomes that are sensitive enough to reflect the biological basis of progressive disease via a measurable clinical endpoint.
 FIGURE 1: MS clinical profile may result from different pathological axes. Two patients with the same clinical profile driven by different pathologic mechanisms. Patient 1: underlying disease is characterized by chronically active lesions and axonal loss; Patient 2: prominent cortical demyelination is driven by meningeal inflammation and neurodegeneration. Indented axes indicate onset of pathology at relatively later stages. Figure adapted from Pitt et al. [3▪▪].
FIGURE 1: MS clinical profile may result from different pathological axes. Two patients with the same clinical profile driven by different pathologic mechanisms. Patient 1: underlying disease is characterized by chronically active lesions and axonal loss; Patient 2: prominent cortical demyelination is driven by meningeal inflammation and neurodegeneration. Indented axes indicate onset of pathology at relatively later stages. Figure adapted from Pitt et al. [3▪▪].In their call for a mechanism-driven framework for characterizing the MS disease course, Kuhlmann et al.[16] note that these pathologic processes occur against the inexorable impact of aging, which is characterized by decreased resilience and increased susceptibility to injury across the disease course [2▪▪,17]. This approach frames MS as a spectrum of overlapping pathological and reparative mechanisms (to different degrees) modified heterogeneously by important negative and positive factors (e.g. lifestyle behaviors, comorbidities, social determinants of health, and disease-modifying treatment) to yield an individual's disease. This perspective is congruent with work on factors that enhance or deplete functional reserve [18] and echo the hypothesis framed by the topographical model of MS, that clinical disease manifestations in an individual patient are likely driven by the extent to which reserve compensates for CNS damage [19].
Furthermore, while specific genetic markers and protein signatures have not yet been fully realized in MS [20], a genetics-informed approach in MS may ultimately enable us to better determine not only who is at risk of developing MS, but the genetics of resilience, to further prognosticate individual expressions of disease [21▪▪].
Although the “ground truth” of an individual patient cannot be directly measured, and can only be gleaned by inference, the concept of precision medicine in MS intends to narrow this gap. It will also help to reduce the use of chameleon concepts such as “smoldering MS” [22] which, while omnipresent in the discourse, can be subjectively interpreted as encompassing various pathologic processes and thus mask the specificity needed to target the disease mechanistically. In “A roadmap to precision medicine in MS,” Chitnis et al.[10] emphasize the importance of focusing on the intrinsic biology underlying MS as a means for truly classifying disease versus anchoring to clinical presentation alone. This will have implications for the development and approval of novel interventions, which in the future will likely need to demonstrate an effect on molecular targets in addition to clinical outcomes. A move toward greater precision in MS has multiple benefits from prognostication of disease – perhaps informed by genotyping – to tailoring of interventions to address specific mechanisms. Indeed, as Khulman et al. also suggest, there is the need to identify and quantify the impact of mechanisms on an individual level via studies that correlate clinical, patient-reported, radiological, and pathological measures. To do so, studies will need to be undertaken in populations without evidence of disease activity to enable better assessment of underlying nonlesional mechanisms using multimodal metrics, such as advanced imaging, paraclinical tools, and more sensitive clinical measures.
2. Emerging biomarkers for disease mechanisms Laboratory assaysThe concept of “ground truth” can only be assessed inferentially through laboratory and imaging metrics that reveal biological processes at the individual level. Identification of such biomarkers specific for particular pathways, processes, and structures are an emerging area of research [3▪▪]. Neurofilament light chain (NfL) remains the most promising marker for MS-related CNS injury, though it is highly variable across individuals. NfL reflects neuroaxonal loss [23], can be assayed in the serum and CSF, and is predictive of new or enlarging T2 lesions and brain volume loss [24▪,25]. Recent data demonstrated elevated sNfL levels even prior to the development of the first MS symptom, with further increases after diagnosis [26].
NfL levels correlate well with disease activity, however data on the association with disability accrual has yielded conflicting results. A large cohort study from two observational cohorts provides insights about the contribution and timing of neuroaxonal injury preceding disability events, with sNfL elevation detected in advance of clinical worsening associated both with, and independent of, relapses [27▪]. This has important implications for understanding not only the mechanisms contributing to disability, but the timeframes in which to potentially target the pathology.
Other biomarkers being studied include those associated with astrocyte activation such as glial fibrillary acidic protein (GFAP) [28] and glycoproteins associated with activated microglia and macrophages, e.g., chitinase [29] and osteopontin [30]. Pitt et al. suggest that the most viable approach may be to develop measures of activation of certain cell types and to consider doing so with a combined approach to best capture distinct patterns of disease. Despite recent investigations, such laboratory assays are not yet actionable in clinical practice, and lag behind imaging biomarkers.
Novel imaging techniquesAdvanced imaging techniques provide increasingly valuable insights into the spectrum of disease in MS. Novel metrics have already evolved the concept of MS from that of a white matter disease to revealing the prognostically-important regions of grey matter pathology [31,32]. Recent studies elucidated the important role of cortical lesions, which are now included in contemporary MS diagnostic criteria [33]. These contributions include the recent understanding of the extent of meningeal inflammation that occurs at or near the site of subpial cortical demyelination. Cortical inflammation has also been shown to have a strong correlation with clinical outcomes [34▪].
Emerging imaging techniques have begun to catch up with the neuropathologists’ view, with the ability to image a wide range of prognostically-relevant tissue damage [11▪].
There has been a heightened focus on chronic active lesions (CALs) which are now understood to occur throughout the disease continuum [35,36]. Slowly expanding lesions (SELs) correlate with greater disability, worse cognition, and progression [37,38▪▪,39]. A recent study indicated that 30% of T2 lesions are SELs, and that almost all patients have at least 1 SEL [38▪▪].
Paramagnetic rim lesions (PRLs) are comprised of a dark ring thought to represent deposition of iron within macrophages and/or activated microglia. PRLs are fewer in number than SELs, but have also emerged as an indicator of poor prognosis. Just over half of patients in a recent study had greater than 1 PRL [38▪▪]. Recent studies showed that lesion expansion and activated rims are not always seen together, underscoring how these techniques may reflect different pathological processes and may vary in their timing during the disease continuum [37,38▪▪]. Co-location of PRLs and SELs have been shown to synergistically contribute to disability increase [38▪▪].
Though parenchymal volumetric techniques are not new, the measurement of brain atrophy remains incompletely adopted in clinical practice due to challenges with reproducibility. Brain volume loss has long been observed from the earliest timepoints in the disease continuum [40] and occurs across different regions of the CNS [41]. Both grey and white matter volumes can be significantly reduced in people with RIS [42–44]. Increasing evidence suggests that the thalamus may be implicated in the early degeneration in MS [45,46]. In a foundational case-controlled study, mean thalamic volumes were found to be significantly lower in RIS patients versus controls [42]. These results can be interpreted to suggest that RIS may not be the “earliest manifestation of the disease” as it is often considered, but may reflect a longer duration of underlying disease processes that had transpired without crossing the threshold of clinical detection. Recent work has identified cervical cord atrophy as a predictor of disability accumulation [47▪▪,48▪]. In aggregate, these data support extensive loss of parenchymal volume driving the decline of the clinical threshold throughout the disease course [7], providing an imaging argument for the overlap between focal inflammatory and diffuse neurodegenerative processes rather than a disease characterized by discrete stages [40].
Considering emerging MRI techniques to visualize the spectrum of MS, one can envision how a pathology-informed imaging assessment framework can further refine our approach to diagnostic [49,50▪] (e.g., Central Vein Sign [CVS], PRLs), prognostic (e.g., PRLs and MRI measures of atrophy including whole brain, substructure, and spinal cord), and disease monitoring for nonrelapsing progressive disease biology [36,51,52▪] (e.g., PRLs, SELs, PET) biomarkers across the spectrum of disease [52▪].
3. Conventional MRI measures of disease burdenAlthough emerging MRI techniques reveal how much of the disease pathology we are not yet able to visualize in routine practice, conventional MRI measures as used in practice and clinical trials remain a reliable window into mechanisms associated with acute injury for diagnosis, prognosis, and monitoring of individuals with MS [53▪].
Two identical T2 lesions may conceal substantive differences in pathology that only emerging MRI techniques can reveal; the absence of cortical lesions on a 3 T MRI may be undone by the plethora of such lesions revealed on a high field MRI at 7 T. Lesion localization, readily apparent on conventional MRI, is however an essential contributor to the heterogeneity of clinical symptoms and the spectrum of disease severity. Early lesions in either the infratentorial region or spinal cord have been shown to increase the risk of disability progression [54]. As Keegan et al.[55] showed, even a single “poorly placed” lesion in an eloquent location – often in the upper cervical cord – predicts worse outcomes. As depicted in the topographical model, lesion topography is an important predictor of disability progression, with lesions in the “shallow end” of reserve conferring hallmark motor and sensory deficits characteristic of MS [7] (see Fig. 2). These results were replicated in a recent study that demonstrated the impact of early spinal cord lesions on the risk of disability progression as measured by EDSS, as well as added performance measures with greater sensitivity such as the timed 25-foot walk (T25FW) and 9-hole peg (9HPT) tests (EDSS-plus) [56].
 FIGURE 2:
FIGURE 2: Conceptualizing disease course using the topographical model. Three timepoints in MS disease course (Panels A–C) as visualized using the topographical model of MS. The blue water represents functional reserve; those lesions that cross the threshold are clinically apparent. Reserve is a fluid construct, subject to the influence of modifiable factors both positive (healthy lifestyle choices, effective DMT) and negative (cardiovascular and metabolic comorbidities, smoking) that may modulate up and down the level of functional reserve. In Panel A, lesions form as topographical peaks below the clinical threshold; no clinical disability has yet occurred; the MRI shown at right depicts brain volume at the outset of disease. In Panel B, after years of disease, additional lesions have formed in characteristic MS regions (spinal cord, infratentorial, cerebral hemispheres), some of which have crossed the clinical threshold. The disability curve shows (in pink) relapse associated worsening (RAW) – an acute increase in disability from a relapse event with incomplete recovery. Brain MRI at right shows atrophy increasing, driving the decline of the clinical threshold. In Panel C, after several additional years the water level has continued to fall, revealing more burden of disease above the clinical threshold. This gradual accrual of disability is denoted in the clinical disability tracing (in purple) as progression independent of relapse activity (PIRA). Brain MRI at right shows extensive atrophy indicative of the loss of compensatory reserve. In total, the layering of RAW and PIRA depicts how an individual patient may develop disability, accruing through different mechanistic pathways occurring at different points in time over their disease continuum.
In Fig. 2, we utilize the topographical model of MS to explicate the dynamic relationship between factors driving disability progression over the MS continuum. This visualization depicts standard of care assessments of the burden of disease radiologically below the clinical threshold, and clinically apparent above it. The clinical-radiologic paradox that underscores the mismatched heterogeneity between MRI appearance, clinical severity, and uncertain prognosis is explained by the ability, or lack thereof, of compensatory reserve to keep much of the disease burden “submerged” below the threshold.
The topographical model of MS hypothesizes that progression emerges as an unmasking of prior damage as reserve is lost, incrementally revealed above the declining clinical threshold. This is most pronounced in the eloquent CNS structures, i.e., the spinal cord and infratentorial region in the “shallow end” of the CNS where there is less compensatory reserve.
Unfortunately, spinal cord imaging in MS remains all but absent from clinical trials and of suboptimal quality in clinical practice. As noted in a recent viewpoint on the case for advancing spinal cord imaging in MS, these most prognostically-important MS lesions may still be hiding in plain sight, just below our limit of radiographic detection [57].
4. Looking below the traditional clinical thresholdIt is important to note that the concept of a clinical threshold is predicated on the sensitivity of the clinical assessments utilized. Neither conventional imaging nor traditional clinical assessments are adequate to identify deficits traditionally considered “invisible” but now understood to be highly disabling for patients, including cognitive dysfunction, language ability, and fatigue. Work by Lebrun et al. over a decade ago showing that cognitive impairment is observed at an early stage of MS (as early as RIS and CIS) has proved to be prescient [58]. Such findings have been replicated in a recent prospective cohort of patients with lesions characteristic of MS (i.e., RIS) who underwent MRI of the brain and cervical spinal cord and cognitive assessments using the Minimal Assessment of Cognitive Function in MS (MACFIMS), with deficits seen in cognitive performance testing [59]. MS may be only “radiologically isolated” if cognitive and other clinical assessments are unchallenging.
What of patients who might appear normal on exam, but may have early as yet undetected changes in function? A study at Mount Sinai evaluated patients from the RADIEMS cohort of early MS with an Expanded Disability Status Scale (EDSS) score of 0 – defined as neurologically normal – and compared them to healthy controls, finding that more challenging tasks of balance and coordination were able to detect “subthreshold” deficits [60]. Clinical phenotyping may not only lack specificity for pathologic mechanisms of disease, but may also be predicated on assessments lacking sensitivity to both cognitively and physically disabling manifestations. Using challenging tasks and patient reported outcomes may allow us to peer below the traditional clinical threshold and garner a more thorough and personalized sense of the burden of disease below. The concept of “silent progression” – the insidious accrual of disability often thought too subtle to discern [61] – is only silent if we are not listening carefully enough. And the “invisible symptoms” of MS are only invisible if we are not looking sufficiently closely [60].
For these reasons, there is a move toward including patient reported outcomes (PROs) and performance-based measures in clinical practice and research, which are especially useful in assessing function and identifying deficits [62,63]. Performance-based measures can be readily incorporated into the clinical visit and are being utilized in clinical trials for composite endpoints in various combinations, under the rubric of “EDSS plus” [63,64▪,65–67]. Integrating performance-based instruments along with novel digital and discussion tools [66,68] to ensure that patients’ experiences of daily function across domains are centered in the clinical visit will help to better challenge the clinical threshold on exam.
5. Conventional neurological exam/Expanded Disability Status ScaleAs we consider the spectrum of disease and current concepts of disease course, the coarseness and insensitivity of our standard measure of MS disability used in clinical practice and clinical trials confers methodological limitations. If an EDSS of 0 is not, in fact, “normal”, then the entire scale right from the beginning is miscalibrated [60]. The EDSS, based on its component functional systems, remains the standard disability measure used in MS clinical trials. Yet, there are concerns that the scale is not granular enough to capture early changes in function, especially in certain domains. Additionally, while the neurological exam is an excellent tool for assessing physical deficits, it does not account for maximal function at an individual level and may not be sensitive enough to capture early changes in function.
The functional system scores (FSS) are the MS-specific formalization of the neurological exam, and at the individual-patient level, they do intrinsically incorporate localization. Pivotal trials of disease-modifying therapies, however, have used as their endpoints disability metrics based on EDSS scores alone, and incremental changes therein to establish confirmed disability progression (CDP), worsening (CDW), or improvement (CDI). By consolidating disability assessments from the EDSS functional systems into the one top-line EDSS score, we forgo localization, and the individualized expressions of the disease are reduced to a single number. All of the heterogenous mechanisms of disease considered in the previous levels of explanation are reduced to these metrics when evaluated as “predictors of disease progression.” Even the clinical trial gold standard of success – no evidence of disease activity (NEDA) – is predicated on this clinical measure, and that of conventional MRI [69].
How much is being missed below the threshold of both clinical and radiologic detection? Given the pathologic heterogeneity and the coarseness of our tools, have we set the bar of success too low, while we have also not yet developed more personalized outcomes?
One path forward is to leverage real world evidence from observational studies to enable inclusion of more heterogenous cohorts, particularly those from underserved populations not previously included in clinical trials and the rising population of MS patients over the age of 60 [70▪]. Only then can we begin to truly understand how health disparities and poor social determinants of health and age-related comorbidities impact clinical outcomes in MS [70▪,71–74].
6. How disability accumulates: disease trajectory modelingModeling and predicting MS disease trajectory takes a further step of abstraction by extrapolating from EDSS-based measures, with all of their limitations, to yield probabilistic estimates of disability accumulation patterns across MS populations. An increasingly accepted perspective in the field is that the clinical course of MS can be distilled into disability accumulation occurring in two ways: as a result of relapses (RAW, relapse-associated worsening) and in the absence of relapses (PIRA, progression independent of relapse activity). It is also worth recognizing that two of the most pervasive and disabling symptoms in MS are fatigue and cognitive dysfunction – neither of which fits neatly into categorization as a relapse or progression, particularly when based on EDSS measures. Still, the substantial contribution of PIRA has been demonstrated in multiple contemporary studies in large, well characterized cohorts [75,76▪▪–78▪▪]. Taken together, these data support the idea that for a considerable number of patients, disability accumulates insidiously from disease onset—a clinical correlate of the hypothesized loss of reserve in the topographical model and congruent with radiographic evidence of diffuse parenchymal volume loss starting early in the disease. In Fig. 2 Panel B, we depict the accumulation of disability through RAW (in pink), the enduring consequence of a relapse with incomplete recovery. In Panel C, PIRA accumulates gradually (in purple) as reserve declines and brain atrophy proceeds.
The Barcelona group [76▪▪] studied a cohort of patients with a first demyelinating event at baseline, and revealed that one-third of all patients with PIRA had their first disability event within 5 years of symptom onset. Notably, the authors stated that early MS patients with PIRA should be considered to have progressive MS, whether or not they have inflammatory activity and independent of their disability score or disease duration.
Similar findings were reported by the Italian group [77▪▪] in a multicenter, observational, retrospective cohort study in which 40% of the cohort at baseline were early in the clinical course, having had only a first clinical event. In this analysis, a large proportion of confirmed disability accumulation occurred during the first five years of follow-up, with 47.8% of first PIRA and 61.8% of first RAW events occurring during years 1–5 of follow-up. Notably, PIRA was the dominant driver of disability after year 1. The authors conclude that the study findings support the view of MS as a disease continuum where RAW and PIRA occur from the earliest phases of the disease.
An analysis of a large cohort of over 27 000 people with MS enrolled in multiple studies across the Novartis-Oxford clinical trial dataset demonstrated that relapses contribute to disability progression via RAW early in the disease course and PIRA begins early and becomes the dominant drier of disability accumulation as the disease evolves [78▪▪]. DMTs delayed disability accrual by years with the highest potential for delaying disability early in the disease, while preexisting disability and older age were the principal risk factors for further disability accumulation. In this analysis, PIRA was observed in the majority of patients classified by phenotype as having secondary progressive MS (SPMS) and primary progressive MS (PPMS), but also in nearly half of those with relapsing remitting MS (RRMS).
7. Traditional clinical course phenotypesWith the above example of PIRA and RAW measured across RRMS, SPMS, and PPMS, we arrive at the highest-order level of explication of MS disease course, the clinical phenotypes, which have heretofore been unmentioned in this Current Opinion. At this level of explanation, the MS disease course is distilled into essentially two categories: relapsing and progressive disease.
Four distinct phenotypes were defined in 1996 based on descriptions of clinical course, though neither objective biological support nor the ability to link the clinical course with MRI findings was then possible [79]. For three decades, researchers and clinicians in the field of multiple sclerosis have indexed on these clinical course descriptors as a means to guide clinical trial design, FDA indications for disease modifying therapy, treatment recommendations for individual patients, and as a method of communicating the disease course to patients and their care partners. Revised in 2013, the clinical course phenotypes incorporated the concept that disease activity could co-occur with progression, but maintained the distinction that the RRMS phenotype is one without progressive manifestations [1].
The Mount Sinai group showed a decade ago that it is impossible to determine a precise moment that an individual patient transition from a relapsing to progressive form of MS, and that there is a years-long period of diagnostic uncertainty between RRMS and SPMS [80]. In routine practice, we say a patient has RRMS when RAW is the dominant expression of how disability is being accumulated; we shift to classifying a patient as having SPMS when PIRA predominates. Use of the RRMS vs. SPMS classifier is thus more revealing about the clinician's thought process and local standards (including health system and payor/coverage models for access to DMTs) than it is about the patient's pathophysiology.
Studies seeking to identify biological differences between discrete phenotypes are methodologically limited, as they may be looking for biological differences between categories that were not drawn along biological lines [7]. Working backwards down the levels of explanation delineated in this review, it is evident from the prevalence of PIRA in CIS and early RRMS that there is an overlapping spectrum of relapse and clinical progression from the outset of the disease.
Conceptualizing “conversion to SPMS” as a long-term study endpoint belies the pathological and clinical overlap of relapse and progression across the MS continuum. Koch, et al. suggest in a recent editorial on reliability of outcome measures in SPMS trials, that as we are revisiting our biological hypotheses for treatment of progressive MS, perhaps the time has come that we should also revisit the instruments that we use to examine their efficacy [81,82▪]. Indeed, if progressive mechanisms start early, and MS is a disease spectrum, then all RRMS trials are SPMS trials if analyzed carefully enough.
CONCLUSIONAll forms of categorization must forgo nuance in favor of order. What the phenotype categories lack in terms of representing MS as a spectrum of disease, they gain in their clarity as an organizing principle and a communication tool. The recent calls to develop a mechanistic framework for characterizing MS emphasize that we are at a pivotal point in MS therapeutic development where we need to focus on the nuances. This will involve elucidating the relative contributions of overlapping pathological and reparative or compensatory processes that yield disease worsening [2▪▪]. One step toward linking above-threshold clinical course descriptors with disease mechanisms may be to consider worsening disease-whether by RAW or PIRA – more dynamically, over set intervals of time, rather than via fixed phenotype definitions [83▪].
The challenge remains to develop methods to identify and quantify disease mechanisms on the individual patient level, and incorporate the relevant measures into both clinical trials and clinical practice. This is needed for a precision medicine approach in MS, particularly as it relates to tailoring treatment decisions to disease mechanisms indicated by radiologic and serologic biomarkers. As Kuhman et al.[2▪▪] point out, such a personalized approach will require correlative clinical–radiological–pathological studies of people different rates of disease worsening, and over set time intervals [83▪], in essence to create a unified conceptualization of the disease spectrum that bridges the different levels of explanation.
At present, however, the relationships and cross-talk between these levels of explanation are incompletely defined, and the congruence between metrics used at different levels of inquiry have not been determined. There is clearly a mechanistic lag between pathological processes and the imaging, subclinical, and overt clinical manifestations of the disease – and we cannot have a truly mechanistic framework until these have been worked out. Until such time, false equivalencies and unsupported assumptions will get us no closer to understanding how “ground truth” mechanisms inform the clinical expression of the disease. That is to say, we need precision in both our science and our terminology. Microglial activation does not simply equate to phase rim lesions; cortical lesions do not map perfectly onto leptomeningeal follicles. And when we talk about MS progression – and aren’t we always?—we are not speaking in the lexicon of specific cellular mechanisms or individual MRI metrics; rather, the word progression retains its original meaning: the steady and unrelenting accumulation of clinical disability, heterogenous in both cause and consequence. We must bring commensurate nuance and rigor to our assessments of progression in order to ensure that our interventions can meaningfully address it.
AcknowledgementsThe authors are grateful to Dr Samantha Epstein for the invitation to contribute to this issue of Current Opinion in Neurology, and to Dr David Pitt for granting permission to use his figure in this manuscript.
Financial support and sponsorshipNo specific funding is reported for this manuscript.
Conflicts of interestS. Krieger reports consulting or advisory work with Baim Institute, Biogen, Cycle, EMD Serono, Genentech, Novartis, Octave, Genzyme/Sanofi, and TG Therapeutics, and nonpromotional speaking with Biogen, EMD Serono, Genentech, and TG. Grant and research support from Biogen, BMS, Novartis and Sanofi. K Cook is an employee of Heartbeat/Publicis Health, which has received fees from Genentech for medical communication services. C Hersh has received speaking, consulting and advisory board fees from Genentech, Genzyme, Biogen, Novartis, EMD-Serono, Bristol Myers Squibb and TG Therapeutics. She has received research support paid directly to her institution by Biogen, Novartis, Genentech, Patient-Centered Outcomes Research Institute (PCORI) and NIH – NINDS 1U01NS111678-01A1 sub-award. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
REFERENCES AND RECOMMENDED READINGPapers of particular interest, published within the annual period of review, have been highlighted as:
▪ of special interest
▪▪ of outstanding interest
REFERENCES 1. Lublin FD, Reingold SC, Cohen JA, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology 2014; 83:278–286. 2▪▪. Kuhlmann T, Moccia M, Coetzee T, et al. on behalf of the International Advisory Committee on Clinical Trials in Multiple Sclerosis. Multiple sclerosis progression: time for a new mechanism-driven framework. Lancet Neurol 2023; 22:78–88. 3▪▪. Pitt D, Lo CH, Gauthier SA, et al. Toward precision phenotyping of multiple sclerosis. Neurol Neuroimmunol Neuroinflamm 2022; 9:e200025. 4▪. Lublin FD, Krieger SC. MS becomes a treatable disease: 30 years later. Mult Scler 2023; 29:789–792. 5. Insel T, Cuthbert B, Garvey M, et al. Research domain criteria (RDoC): toward a new classification framework for research on mental disorders. Am J Psychiatry 2010; 167:748–751. 6. Kalincik T, Sormani MP, Tur C. Has the time come to revisit our standard measures of disability progression in multiple sclerosis? Neurology 2021; 96:12–13. 7. Krieger SC, Cook K, De Nino S, Fletcher M. The topographical model of multiple sclerosis: a dynamic visualization of disease course. Neurol Neuroimmunol Neuroinflamm 2016; 3:e279. 8. Laitman BM, Cook K, Fletcher M, Krieger SC. The topographical model of MS: Empirical evaluation of the recapitulation hypothesis. Mult Scler J Exp Transl Clin 2018; 4:2055217318806527. 9. Krieger SC, Sumowski J. New insights into multiple sclerosis clinical course from the topographical model and functional reserve. Neurol Clin 2018; 36:13–25. 10. Chitnis T, Prat A. A roadmap to precision medicine for multiple sclerosis. Mult Scler 2020; 26:522–532. 11▪. Yong HYF, Yong VW. Mechanism-based criteria to improve therapeutic outcomes in progressive multiple sclerosis. Nat Rev Neurol 2022; 18:40–55. 12. Kuhlmann T. Relapsing-remitting and primary progressive MS have the same cause(s): the neuropathologist's view: 2. Mult Scler 2013; 19:268–269. 13. Lassmann H. Relapsing-remitting and primary progressive MS have the same cause(s): the neuropathologist's view: 1. Mult Scler 2013; 19:266–267. 14. Dutta R, Trapp BD. Relapsing and progressive forms of multiple sclerosis: insights from pathology. Curr Opin Neurol 2014; 27:271–278. 15. Antel J, Antel S, Caramanos Z, et al. Primary progressive multiple sclerosis: part of the MS disease spectrum or separate disease entity? Acta Neuropathol 2012; 123:627–638. 16. Musella A, Gentile A, Rizzo FR, et al. Interplay between age and neuroinflammation in multiple sclerosis: effects on motor and cognitive functions. Front Aging Neurosci 2018; 10:238. 17. Azevedo CJ, Cen SY, Jaberzadeh A, et al. Contribution of normal aging to brain atrophy in MS. Neurol Neuroimmunol Neuroinflamm 2019; 6:e616. 18. Brandstadter R, Katz Sand I, Sumowski JF. Beyond rehabilitation: a prevention model of reserve and brain maintenance in multiple sclerosis. Mult Scler 2019; 25:1372–1378. 19. Vollmer TL, Nair KV, Williams IM, Alvarez E. Multiple sclerosis phenotypes as a continuum: the role of neurologic reserve. Neurol Clin Pract 2021; 11:342–351. 20. Oksenberg JR, Baranzini SE. Multiple sclerosis genetics—is the glass half full, or half empty? Nat Rev Neurol 2010; 6:429–437. 21▪▪. International Multiple Sclerosis Genetics Consortium, MS Consortium. Locus for severity implicates CNS resilience in progression of multiple sclerosis. Nature 2023; 619:323–331. 22. Giovannoni G, Popescu V, Wuerfel J, et al. Smouldering multiple sclerosis: the ‘real MS’. Ther Adv Neurol Disord 2022; 15:17562864211066751. 23. Khalil M, Teunissen CE, Otto M, et al. Neurofilaments as biomarkers in neurological disorders. Nat Rev Neurol 2018; 14:577–589. 24▪. Sotirchos ES, Fitzgerald KC, Singh CM, et al. Associations of sNfL with clinico-radiological measures in a large MS population. Ann Clin Transl Neurol 2023; 10:84–97. 25. Kuhle J, Kropshofer H, Haering DA, et al. Blood neurofilament light chain as a biomarker of MS disease activity and treatment response. Neurology 2019; 92:e1007–e1015. 26. Bjornevik K, Munger KL, Cortese M, et al. Serum neurofilament light chain levels in patients with presymptomatic multiple sclerosis. JAMA Neurol 2020; 77:58–64. 27▪. Abdelhak A, Benkert P, Schaedelin S, et al. Neurofilament light chain elevation and disability progression in multiple sclerosis. JAMA Neurol 2023; 80:1317–1325. 28. Abdelhak A, Huss A, Kassubek J, et al. Serum GFAP as a biomarker for disease severity in multiple sclerosis. Sci Rep 2018; 8:14798. 29. Martínez MAM, Olsson B, Bau L, et al. Glial and neuronal markers in cerebrospinal fluid predict progression in multiple sclerosis. Mult Scler 2015; 21:550–561. 30. Comabella M, Pericot I, Goertsches R, et al. Plasma osteopontin levels in multiple sclerosis. J Neuroimmunol 2005; 158:231–239. 31. Calabrese M, Magliozzi R, Ciccarelli O, et al. Exploring the origins of grey matter damage in multiple sclerosis. Nat Rev Neurosci 2015; 16:147–158. 32. Eshaghi A, Prados F, Brownlee WJ, et al. Deep gray matter volume loss drives disability worsening in multiple sclerosis. Ann Neurol 2018; 83:210–222. 33. Thompson AJ, Banwell BL, Barkhof F, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol 2018; 17:162–173. 34▪. Magliozzi R, Howell OW, Calabrese M, Reynolds R. Meningeal inflammation as a driver of cortical grey matter pathology and clinical progression in multiple sclerosis. Nat Rev Neurol 2023; 19:461–476. 35. Absinta M, Sati P, Masuzzo F, et al. Association of chronic active multiple sclerosis lesions with disability in vivo. JAMA Neurol 2019; 76:1474–1483. 36. Frischer JM, Weigand SD, Guo Y, et al. Clinical and pathological insights into the dynamic nature of the white matter multiple sclerosis plaque. Ann Neurol 2015; 78:710–721. 37. Elliott C, Wolinsky JS, Hauser SL, et al. Slowly expanding/evolving lesions as a magnetic resonance imaging marker of chronic active multiple sclerosis lesions. Mult Scler 2019; 25:1915–1925. 38▪▪. Calvi A, Clarke MA, Prados F, et al. Relationship between paramagnetic rim lesions and slowly expan
Comments (0)